|
|
本文基于个人VASP应用经验和VaspwikiThe VASP Manual - Vaspwiki官方说明撰写,且仅适用于VASP_v6.3.0版本。
为方便阅读和快速理解,本文采取类似箴言手册的形式撰写。
本文目前包含以下你可能关心的内容:
1)结构弛豫;
2)电子自洽;
3)电子态密度;
4)能带结构;
5)DFT+U;
6)自旋极化;
7)杂化泛函方法计算能带;
8)GW近似方法计算能带;
9)第一性原理分子动力学。
请自行翻阅寻找你所感兴趣的部分。
<hr/>本文内容会被追加于个人文章Centos系统计算化学/材料软件装配方法 & 标准化理论计算与数据处理流程 —— 个人经验记录 - 知乎 (zhihu.com)相应章节中。
You and I just have a dream,
to find our love a place, where we can hide away.
<hr/>关于VASP——The Vienna Ab initio Simulation Package
维也纳从头计算模拟程序包(VASP)是用于原子尺度材料模拟的计算机程序,其基于第一性原理,可以执行电子结构计算和量子力学分子动力学。
VASP能够计算多体Schrödinger方程的近似解,其要么在密度泛函理论(DFT)的框架内求解Kohn-Sham方程,要么在Hartree-Fock (HF)近似内求解Roothaan方程。混合了Hartree-Fock方法和密度泛函理论的杂化泛函计算也可以实现。此外,在VASP中还可以使用格林函数方法(GW准粒子和ACFDT-RPA)和多体微扰理论(二阶Møller-Plesset)。
在VASP中,中心量,如单电子轨道、电子电荷密度和局域势都以平面波基组表示。电子和离子之间的相互作用以模守恒赝势、超软赝势或缀加投影平面波方法来描述。
为确定电子基态,VASP使用高效迭代的矩阵对角化技术,如带有迭代子空间直接反演的残差极小化方法(RMM-DIIS)或阻塞Davidson算法。这些算法与高效Broyden和Pulay密度混合方案相耦合,用以加快自洽循环。
更多关于VASP的详细介绍请参考VASP - Vienna Ab initio Simulation Package。
计算准备与基础认知
- 开启一个VASP计算任务需要事先准备四个基本输入文件:POSCAR、POTCAR、KPOINTS和INCAR。
- POSCAR是包含目标体系结构信息的文件。第一行是说明信息,无所谓。第二行是晶格缩放系数,有些晶格优化方法会用到。三到五行是晶格参数。第六行是元素符号,有没有这行不影响VASP计算,但加上为好。第七行是各元素原子数。第八行指定原子的坐标形式,有笛卡尔坐标(C)和分数坐标(D)。第九行开始就是各原子的坐标。
- 固定原子坐标时,要在原第七和第八行间增加一行内容:Selective Dynamics,并你在所有你要固定的原子坐标后面增添三个F,在不固定的原子坐标后面增添三个T。
- POSCAR通常转化CIF文件而来,VESTA和VASPKIT是两个很方便的工具程序。
- POSCAR文件中元素的顺序非常重要。POSCAR、POTCAR、INCAR文件中的内容或设置都涉及到元素的顺序,谨记要保持它们的顺序一致。
- POTCAR是赝势文件,你需要自己决定用哪种赝势进行你的计算。你可以用shell—cat命令将选用的赝势文件合成一个POTCAR文件,不过VASPKIT可以很方便的实现这一功能。赝势的选取同时决定了计算所使用的泛函(不需要在INCAR中设置,VASP会默认使用赝势文件中指定的泛函)。VASP使用的赝势通常都属于PAW类型。GGA-PBE是VASP计算最常用到的泛函。同一个元素可能有很多种赝势,它们分别代表考虑不同价电子数的情况,你可以用shell—vim命令查看赝势的内容。GW计算需要用到带有_GW后缀的赝势。
- KPOINTS是关于K点设置的文件,计算任务的类型不同,KPOINTS的内容不同。VASPKIT可以很方便的帮你生成你所需要的KPOINTS文件,不过你完全可以自行建立一系列KPOINTS模板,因为它包含的内容并不多。
- INCAR是关于VASP计算任务参数设置的文件,也是最需要精心准备的文件。VASPKIT可以帮你快速生成你所需要的INCAR文件,但是最好还是自己设置,因为用默认参数值或第三方软件帮你设置好的参数值进行计算往往达不到你想要的效果,也不够灵活。如同KPOINTS文件一样,你完全可以自行建立一系列适合你体系和计算任务的INCAR文件。
- INCAR文件内容我们可以分成四个部分看待:1)基本参数设置;2)SCF电子自洽;3)目标任务部分;4)额外补充部分。
- INCAR文件的基本参数设置部分通常涉及到如下标签(tags):
SYSTEM = 体系名称 #给你的体系起个名字吧.无所谓,但如果你不设,你会发现输出文件默认命名为unkonwn.
ISTART = 0 或 1 #读(1)不读(0)取WAVECAR.此行根本不需要设置,VASP默认会在存在WAVECAR时读取,不存在就不读取.
ICHARG = 默认2(ICHARG=0)或0 #在做电子态密度和能带结构计算时我们需要读取一步SCF电子自洽计算的CHGCAR,要把此项设为11.其他情况就把此行删掉保持默认即可.
LWAVE = .TRUE. 或 .FALSE. #要不要输出WAVECAR.结构优化不用输出,电子自洽务必输出,因为不论是电子态密度还是能带计算都需要读取一次电子自洽任务输出的WAVECAR或CHGCAR.
LCHARG = .TRUE. 或 .FALSE. #要不要输出CHGCAR.同理如上.
LELF = .TRUE. 或 .FALSE. #要不要输出ELFCAR.如果你想要得到电子局域函数就要将这项打开.注意打开此项时一定保持KPAR默认,否则VASP会BYE.
LORBIT = 默认None,建议设为11 #输出一些额外的信息.这项就直接设成11就好,你可以自此在OUTCAR中看原子磁矩和原子电荷等信息,电子态密度和能带结构计算也需要此项.
NCORE = 默认值1,请参考你机器的硬件配置设置 #控制并行计算优化的参数.老版本VASP是用NPAR控制的,现在用NCORE控制更好.合理的设置该项可以使你的计算加快最高4倍哦.不过谨记非DFT计算不要设置此项,默认就好.注意有时设的过&#34;快&#34;了会出现内存不足问题而使计算BYE,这个时候大不了就&#34;慢&#34;一点.
SYMPREC = 默认值1E-5 #这个控制的是VASP读取POSCAR文件对称性的精度,建议提高一点,如设为1E-4.太低有时候会导致倒空间对称性与实空间不匹配而BYE.
- INCAR文件的SCF电子自洽部分通常涉及到如下标签:
ENCUT = 默认从POTCAR中读取 #切记合理设置此值,不要用默认值.
EDIFF = 默认1E-4 #默认值太低.可以提高到1E-6.有些计算如GW要求更高要到1E-8.
NELM = 默认60 #默认值太低.完全可以大幅提高,如到200.不过VASP官网说的没错,如果你算到60步了还不收敛就要考虑是不是INCAR有问题.
ISMEAR = 默认1 #该项非常重要.一定要具体问题具体分析.一般来说,对绝缘体和半导体取ISMEAR=-5(注意ISMEAR=-5时不需要添加SIGMA)或ISMEAR=0结合SIGMA=0.03-0.05;对金属体系做结构优化取ISMEAR=1或2结合SIGMA=0.2;对任何体系进行准确SCF电子自洽和态密度计算均建议取ISMEAR=-5,但能带结构计算不要这么取.
SIGMA = 默认0.2 #与ISMEAR配合使用.
ALGO = 与计算任务有关 #自洽算法设置.一般来说做DFT计算时设为Normal就好,对某些体系追求速度可以设为Fast或VeryFast,但有时这样设不仅不收敛反而速度更慢.做杂化泛函和GW计算时此项有不同的值.
PREC = 计算精度设置 #官网推荐Normal或Accurate.
ADDGRID = .TRUE. 或 .FALSE. #增强计算计算过程.True不一定有效,但一般打开就行,出错再改.
###结构优化###
IBRION = 优化算法 #一般用2就好.
ISIF = 2或3 #只优化原子坐标用2;既优化原子坐标又优化晶格形状用3.
NSW = 默认0 #最大离子步数.切记一定要自己设一个数如100,默认设置是不跑离子步不做结构优化的.
EDIFFG = 通常设-0.05,-0.02或-0.01 #收敛条件.越严格就把值的绝对值设越小.负值表示用力做标准,最常用.也可以设为正值用能量做标准,不过该设多少就得自己琢磨了. ###电子态密度和能带计算, 两个内容几乎一样但也有区别, 区别见后文###
NBANDS = 默认就好 #能带数量,没有更好的想法就保持默认.GW计算要仔细设这个值.
EMIN = 能量范围下界 #关心到哪就设哪.
EMAX = 能量范围上界 #关心到哪就设哪.
NEDOS = 取点数,默认301 #太小,多一些点作图更平滑,可以设到2000甚至3000. ###第一性原理分子动力学###
IBRION = 0 #表示我要做动力学.
SMASS = -3或-2或-1或大于等于0的实数 #速度控制.正则系综NVT系统请设为一个大于等于0的实数.
ISIF = 2 #只让原子动.
POTIM = 每一步的时长 #动力学一般跑到5ps也就是5000fs,如果你设此值为1那么你就要设NSW为5000.
NSW = 步数 #不解释了.
MDALGO = 2 #动力学方案.有0,1,2,3,11,21,13七种选择.比如研究正则系综NVT系统可设为2,采用Nose-Hoover恒温器方案.同时不要忘了设起始和结束温度.
TEBEG = 起始温度 #热力学温度,单位K.
TEEND = 结束温度 #热力学温度,单位K.
###自旋极化设置###
ISPIN = 2 #设成2开启自旋极化.
MAGMON = 原子磁矩设置,格式:原子数*磁矩 #没有关于原子磁矩的先验知识就默认.
AMIX = 0.2 #下列此项都是控制电荷密度线性混合的参数,在做自旋极化计算时要合理设置,找见对的值才能助你找到你想要的磁性状态.如果不知道设什么好,这里的设置就是官网的推荐值,先试试看.
BMIX = 0.0001
AMIX_MAG = 0.8
BMIX_MAG = 0.0001###DFT+U###
LDAU = .TRUE. #考虑DFT+U.
LDATYPE = 2 #加U方法.2好像最常用.
LDAUL = 加U轨道设置,按POSCAR中的元素顺序设 #对应关系[-1 1 2 3]-[无 p d f].根据你的体系设置.
LDAUU = U值 #根据你的体系设置.按POSCAR中的元素顺序设.
LDAUJ = J值 #同理如上.
LMAXMIX = 4或6 #考虑d电子时设4;考虑f电子时设6.###杂化泛函###
LHFCALC = .TRUE. #开启杂化泛函计算.
AEXX = 默认0.25 #交换部分参数.默认就好.
HFSCREEN = 0.2 #用HSE06.
ALGO = 注意此项与SCF电子自洽重复 #算法.带隙大用All,带隙小用Damped.把SCF电子自洽部分的此项删掉.
PRECFOCK = Normal或Accurate #Normal就行.你会知道杂化泛函计算速度非常慢.###GW计算, 算GW时把SCF电子自洽的部分删掉换成这个###
还在紧张撰写中.
- INCAR文件编写内容时不用拘束格式,VASP会自动忽略含义不清的行。
- VASP输出文件有很多,常用到WAVECAR(波函数信息)、CHGCAR(电荷密度信息)、ELFCAR(电子局域函数)、CONTCAR(输出结构)。动力学会用到XDATCAR(动力学轨迹),有很多后处理程序可以方便的将之转变为xtd、xyz等格式文件,有助于你去做动力学轨迹动画图。电子态密度和能带结构计算会用到vasprun.xml,配合Pymatgen可以快速做出电子态密度和能带结构图。杂化泛函计算会用到IBZKPT用以设置能带计算KPOINTS文件,当然你也可以使用VASPKIT快速生成杂化泛函能带计算要用的K点文件,但自己设更能保持一致性(不同能带结构计算横向对比时)。
通用计算流程
结构优化
- 拿到一个材料的结构肯定先要优化。你可以直接设ISIF=3让原子坐标和晶格形状都动,但官方似乎不建议这么做。官方建议先固定原子坐标做晶格形状优化,然后再用优化好的结构做固定晶格下原子坐标的优化,注意连续计算时用前一步的CONTCAR做后一步的POSCAR.
- 固定原子坐标优化晶格形状时特别是晶格参数时最好的做法就是控制其它参数不变去调整晶格缩放系数,做很多个不同系数下的SCF电子自洽,比较它们的能量,选最低的那个,缩放后的结构就是优化了晶格参数的结构。
- 结构优化不用输出WAVECAR和CHGCAR,注意它们很占你的硬盘空间哦。
SCF电子自洽
- 这部分没什么好说的,Just do it。
- 一定要输出WAVECAR和CHGCAR,这样在你以后不论是计算电子态密度和能带结构,还是做杂化泛函和GW计算,就直接读取该步的结果继续算就行了,免得重复计算。
电子态密度和能带结构
- 都是读取前一步SCF电子自洽的WAVECAR和CHGCAR去算,记得设ICHAGE=11。
- 电子态密度计算KPOINTS设置与SCF电子自洽相同即可。能带结构计算要设置K点路径,VASPKIT是个好工具。
- 使用杂化泛函时只需读取前一步SCF电子自洽计算的WAVECAR,别读CHGCAR,也就是说ICHAGE默认就行,这是因为我们把泛函给杂化了,用到了Hartree-Fork方法。另外,杂化泛函算能带时KPOINTS又有不同,VASPKIT是个好工具,不过自己设也很简单:把SCF电子自洽输出文件IBZKPT中前面关于倒空间的部分复制一下,建个新文件命名KPOINTS,粘贴进去,然后在后面追加K点路径,但要把最后关于高对称点名称的部分删掉都改成0,保存,完事。
- GW计算又有不同,正在紧张撰写中。
第一性原理动力学
最后贴个用Pymatgen画电子态密度和能带结构的python脚本,以防忘记。
### 分波投影电子态密度 ###
import matplotlib.pyplot as plt
from pymatgen.electronic_structure.core import Spin
from pymatgen.electronic_structure.plotter import DosPlotter
from pymatgen.io.vasp.outputs import Vasprun
dosrun = Vasprun(&#34;vasprun.xml&#34;, parse_dos=True)
dos = dosrun.complete_dos
dosplot = DosPlotter(sigma=0.1)
dosplot.add_dos(&#34;Total DOS&#34;, dos)
dosplot.add_dos_dict(dos.get_element_dos()) # 默认画元素投影,将括号内容改为dos.get_spd_dos()就是轨道投影。
plt.rcParams[&#39;xtick.direction&#39;]=&#39;in&#39;
plt.rcParams[&#39;ytick.direction&#39;]=&#39;in&#39;
plt = dosplot.get_plot(xlim=(-3,3),ylim=(0,80))
plt.yticks([])
plt.savefig(&#39;epdos.png&#39;)
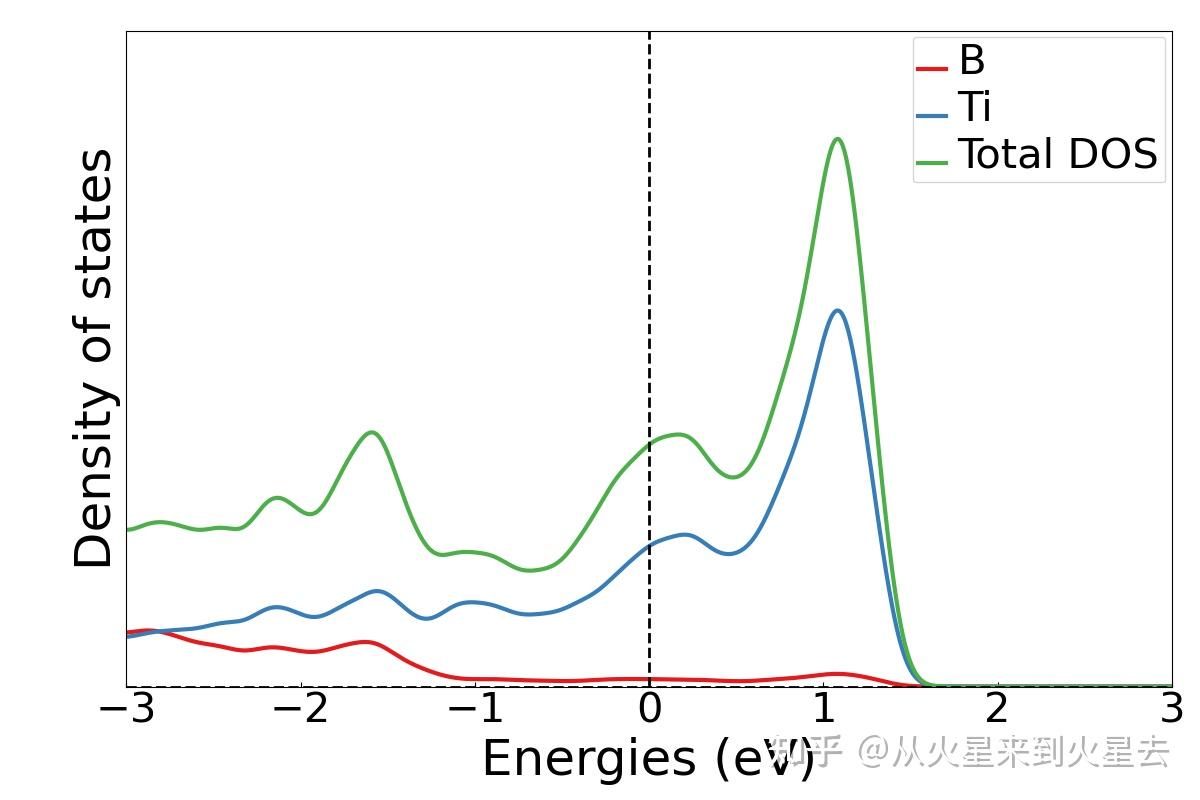
元素投影电子态密度
### 能带结构+分波投影电子态密度 ###
import matplotlib.pyplot as plt
from pymatgen.electronic_structure.core import Spin
from pymatgen.electronic_structure.plotter import BSDOSPlotter, BSPlotter, DosPlotter
from pymatgen.io.vasp.outputs import BSVasprun, Vasprun
run = BSVasprun(&#34;vasprun.xml&#34;, parse_projected_eigen=True)
dosrun = Vasprun(&#34;../dos/vasprun.xml&#34;, parse_dos=True)
dos = dosrun.complete_dos
bs = run.get_band_structure(&#34;KPOINTS&#34;, efermi=dos.efermi)
bsdosplot = BSDOSPlotter(bs_projection=&#34;elements&#34;, dos_projection=&#34;orbitals&#34;, vb_energy_range=3, cb_energy_range=3, fixed_cb_energy=3, fig_size=(9,6))
plt.rcParams[&#39;xtick.direction&#39;]=&#39;in&#39;
plt.rcParams[&#39;ytick.direction&#39;]=&#39;in&#39;
bsdosplot.get_plot(bs=bs, dos=dos)
plt.savefig(&#39;bdos.png&#39;)
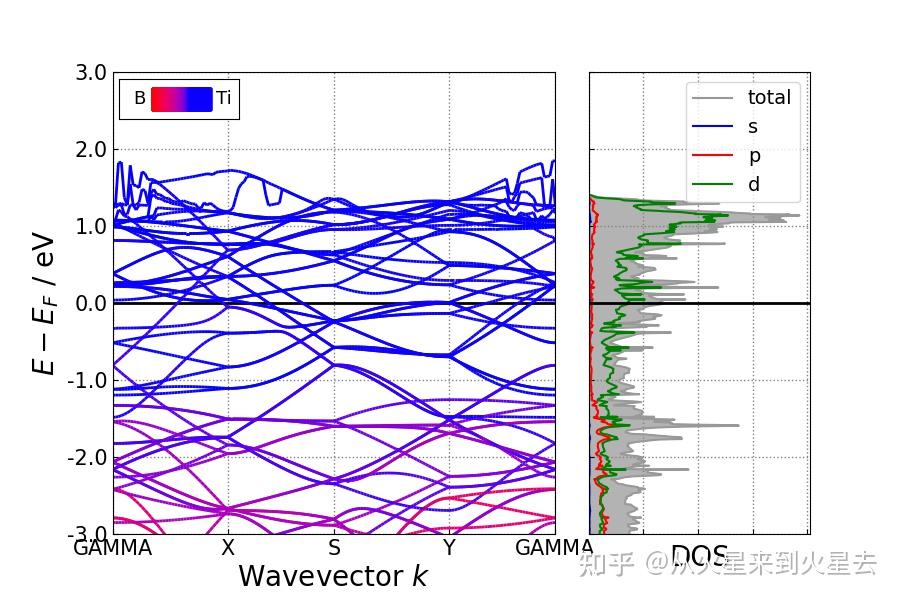
能带+DOS
求点赞并关注。 |
|



 发表于 2022-11-14 13:56:44
发表于 2022-11-14 13:56:44